Page 1, 2, 3, 4, 5
The pathological effect of microcirculatory protein glycation is a stiffening of the capillaries throughout the affected organ – actually a caramelization of the microvascular skeleton within the organ involved. This process involves both abnormal biochemistry (protein glycation) and structural circulatory regulation changes (increased microvascular resistance). The gradual glycation of protein molecules ("endothelium") lining the small capillary microcirculation stiffens those microvessels so that they cannot pulsate with the heartbeat. In addition, glycation of the proteins on the red blood cell stiffens their external membranes, making it more difficult for the stiffened red cells to pass through the caramelized capillary beds one at a time. The resultant structural problem is the inability of the microcirculation to pulsate open with the kinetic (pumping) force of the heartbeat, coupled with the red blood cells' inability to bend, twist, and deform to slip through these stiffened capillaries one at a time. A capillary that is pulsed open during cardiac systole is 10 microns in diameter. During diastole, the capillary reduces to 5 microns in diameter. A red blood cell is 8 microns in diameter. Thus, the caramelized capillary cannot dilate effectively, and the older caramelized red cell has reduced flexibility. This situation results in small capillary "infarcts or strokes" in the affected microcirculatory vessel directly involving the affected organ or tissue (i.e., brain, heart, kidney, etc.).
Over time (years to decades) the collective effects of this process appears clinically. Interestingly, the clinical syndrome that appears depends on which organ(s) is (are) affected. Microcirculatory disease somewhat mimics macrocirculatorydisease in that it may "skip" around, affecting different organs or areas of the body differently. Thus, if microcirculatory glycation occurs in the heart there will be a development notof a major clinical "heart attack" (myocardial infarction), but small areas of tissue damage (i.e., slight elevation of enzymes without ECG changes) or, more importantly, the gradual development of diastolic heart failure.65-82 This process does not involve the active process of the heart contracting but its inability torelax effectively (diastole) after contraction due to microvascular stiffening of capillaries within the involved heart muscle. Thus, the heart fails to fill effectively and clinical congestive heart failure (CHF; fluid backup into the lungs, legs, etc.) develops. CHF is the now the most common and least curable form or heart disease affecting the American population, with a large proportion being diastolic heart failure. Similarly, if capillary glycation involves primarily the kidney, high blood pressure will develop. Interestingly, diastolic dysfunction, diastolic heart failure, high blood pressure, and sugar toxicity ("metabolic syndrome") are now "epidemic" among Americans, old and young.83,84 If capillary glycation occurs mainly in the brain, a condition called leukoaraiosis or coalescing tiny (lacunar) strokes leading to scaring of the gray matter in the brain occurs, and memory loss/cognitive dysfunction and/or small strokes (lacunar infarcts) will develop.79,80 Leukoaraiosis is still being taught in scientific medical schools as "a result of aging" and is of "no medical consequence." This outdated thinking has been clearly and scientifically disproved. The presence of leukoaraiosis on an MRI or CT brain scan should be taken as direct clinical evidence of abnormal microvascular glycation (endothelial dysfunction).85-128 The connection between heart disease, hypertension, and Alzheimer's syndrome, suggesting a common underlying mechanism (microvascular disease), is now being formally recognized.129-134
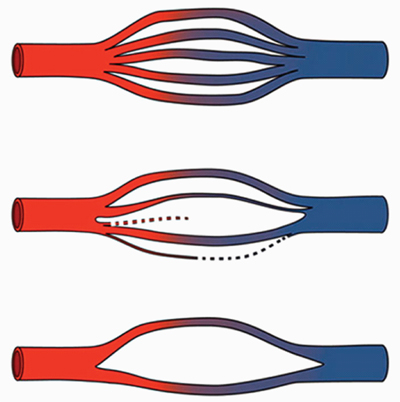
Figure 1 depicts a diagrammatic representation of this gradual process of capillary cross-sectional attrition over time from normal capillary flow through a tissue or organ in the upper diagram to an intermediate reduction in the middle and then finally a significant loss of the illustrated microvascular capillary bed on the bottom. The organ or tissue most affected is where the clinical "syndrome picture" will manifest (heart, kidney, brain, etc.).
Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011 Feb;37(1):64.
Reprinted with permission.
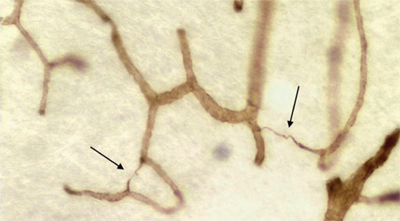
Figure 2 is a photomicrograph of a section of brain clearly demonstrating the loss of the microcirculatory capillary bed in a histologic section of brain from a patient with dementia.
Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011 Feb;37(1):61.
Reprinted with permission.
Interestingly, in addition to the toxicmicrocirculatory effects of glucose and subsequent endothelial glycation now being described in Alzheimer's-like senile dementia, scientific evidence also shows that the accumulation of abnormal proteins (β-amyloid and tau protein) in Alzheimer's disease also causes reduced microcirculation (vasoconstriction) in the brain. β-amyloid protein also reduces endothelium-dependent brain vasodilation. As if to add insult to injury, any temporary lack of oxygen in the brain can also lead to increased production of β-amyloid protein. Thus, it is apparent that microcirculatory compromise plays a very large role in the development of memory loss, cognitive dysfunction, and dementia syndromes, regardless of the actual "official name" of the clinical syndrome. In addition to and as further proof of this situation, the microscopic changes in brain tissue that are thought to be "specific" for Alzheimer's disease (β-amyloid, tau protein, neurofibrillary tangles) have also been described in patients with heart vascular disease and no evidence of dementia. The increase in body fat that is part of "metabolic syndrome" also contributes to the "bad" situation. Centralized body fat is actually a functioning endocrine organ. Body fat secretes certain pro-inflammatory cytokine hormones (tumor necrosis factor alpha and interleukin-6, etc.). Thus, increased body fat leads to increased inflammation (a statistical risk factor for macrocirculation disease). Inflammation can be measured using the blood test C-reactive protein or CRP. Thus, many previously recognized "independent risk factors" can now be connected through the understanding of the biologically toxic properties of sugar related to malfunction of cellular insulin receptors.
Insulin Resistance/Metabolic Syndrome
The underlying metabolic basis of glucotoxicity is the inability of the enzyme insulin to activate insulin receptors on cell surfaces ("membranes") that allow the sugar to move quickly from the blood into the cellular metabolism, where it can be used for energy production (and the production of free radicals!). Thus insulin acts as a "key" that must fit into an insulin receptor or "lock" and open that lock so that potentially toxic sugar can be transported ("disposed of") quickly and efficiently from the blood into the cellular metabolism. When sugar is not disposed of quickly, several "bad" things begin to happen: (1) glycated microvessels and cellular red corpuscles "stiffen" and lose their ability to pulsate with the heart; (2) the undisposed-of sugar is immediately shunted into centralized body fat production. One of the clinical markers used to define metabolic syndrome is centralized body fat (a large waistline); (3) additional pathological events related to poor glucose disposal include excess sorbitol production leading to nerve damage (diabetic neuropathy) and cataracts. The "defining components" of metabolic syndrome that have been "suggested" or adopted by various scientific groups and societies are high blood pressure, high cholesterol, high triglycerides, high fasting insulin levels, and high fasting blood sugar. Interestingly, all the different criteria put forth by various scientific medical groups (the WHO, American College of Cardiology, American Association of Clinical Endocrinology, etc.) do not include any evidence of direct tissue sugar-protein glycation as criteria for diagnosis. Usually, the presence of three or more of these statistical risk factors "qualifies" as a diagnosis of "metabolic syndrome." In reality, by the time any individual actually "qualifies" for the formal diagnosis of metabolic syndrome, they are already manifesting significant and late-stage clinical evidence of severe microvascular glycation (i.e., high blood pressure, obesity, heart disease, dementia, etc.).
The main biochemistry involved in the pathology of glucotoxicity is known as the Maillard reaction. This common chemical reaction has been known for over 100 years. The main scientific focus of this reaction usually involves food spoilage and industrial applications. Other than in diabetes mellitus ("sugar diabetes"), there was really little medical scientific interest in the Maillard reaction in medical science or practice.135 However, with the recent and gradually increasing scientific realization that the cholesterol ("fat" toxicity) theory of cardiovascular disease is at best incomplete, or more likely simply wrong, more scientific interest and resources have been reevaluating the pathophysiological effects of the Maillard (sugar toxicity) reaction.136-141 There is also overwhelming scientific evidence rapidly accumulating that in vivo pathological Maillard reactions accumulate both from internal dysmetabolism of sugar and ingesting advanced glycation end products (AGEs) created by excessive heating of food that are toxic in living tissue.142 In addition to the Maillard reaction, the Amadori rearrangement and Schiff base reactions are also part of the complex biochemistry of sugar toxicity.135
One major problem faced by clinicians interested in prevention of and early intervention in the manifestations of abnormal tissue glycation is that there is no single test in "evidenced-based" medicine that can "diagnose" metabolic syndrome. Abnormal glycation diseases can be diagnosed using several clinical and laboratory findings, but not by a single laboratory test. A simple blood test called the hemoglobin A1c or glycohemoglobin (HbA1c) test easily and cheaply provides critical clinical information about the ongoing degree of glycation in the body due to inadequate glucose disposal from blood.143-146 The "trick" in using this scientific (evidence-based) test as a "new and emerging risk factor for clinical vascular disease" (as it is now being discovered and described in scientific medical journals) is to understand that the laboratory statistical "norms" are not related to individual optimal values. A value for HbA1c above 4.6 should be considered "abnormal" (meaning evidence of accelerated cellular membrane glycation). In most commercial laboratories diabetes is said to be present,by definition, if the value is 6.4 or higher. This is why most diabetics demonstrate severe vascular disease before scientific medicine diagnoses it. Membrane receptor resistance to the hormone Insulin is not acknowledged until late in the process (advanced tissue glycation) or a "high" HbA1c. Objectively testing using PORH for disturbed microcirculatory function and/or pulse volume recordings (PVR; microvascular disease of the vasa vasorum in macrovessels) can also demonstrate the effects of abnormal vascular glycation (loss of microvascular compliance).
As an aside regarding laboratory measurement of insulin or any other "hormone level," the clinical reality is that no hormone actually works in the blood (or urine, or saliva); hormones do their metabolic workby activating a hormone receptor (i.e., the "key in lock" analogy) located on a specific cell or nuclear membrane. The hormone receptor is usually located on the cell membrane (i.e., insulin receptor) or the nuclear membrane (i.e., thyroid receptor). Thus, in many individual ("not statistical") cases, the "blood level" (and urine or saliva, for that matter) of a given hormone (i.e., thyroid, insulin, vitamin D, etc.) may be "statistically" normal or even "optimal" and yet the clinical symptoms of the particular hormone "deficiency" may clearly be present. This is a very common clinical phenomenon seen with insulin resistance syndrome. Blood simply acts as a transport medium to get the hormone from where it was made to where it will have its metabolic effect. As stated earlier, the insulin receptor is located on the cell membrane – and some cells have many more insulin receptors than others. This is the functional basis of IPT (insulin potentiation therapy) cancer chemotherapy: to use insulin to activate the insulin receptors on cancer cells. Cancer cells revert to a more primitive cellular metabolism referred to as anaerobic ("without oxygen") glycolysis (fermentation). Thus, cancer cells have significantly more insulin receptors on their cell surface than noncancer cells. Giving insulin to cause a drop in blood sugar, and at the low point of the blood sugar very small (15% to 20% of standard, recommended) doses of chemotherapy drugs to allow the cancer cell to preferentially take in the drugs as they are in their insulin activated sugar "feeding frenzy," creates a physiologic "Trojan horse" approach that significantly minimizes side effects and yet maximizes therapeutic benefit of chemotherapy to actually target the problem cancer cells, thus sparing drug toxicity to normal cells and tissue. The clinical results in cancer can be quite amazing when appropriately employing IPT.
Page 1, 2, 3, 4, 5
|




